Research HighlightsIn Silico Methods for De Novo Protein Design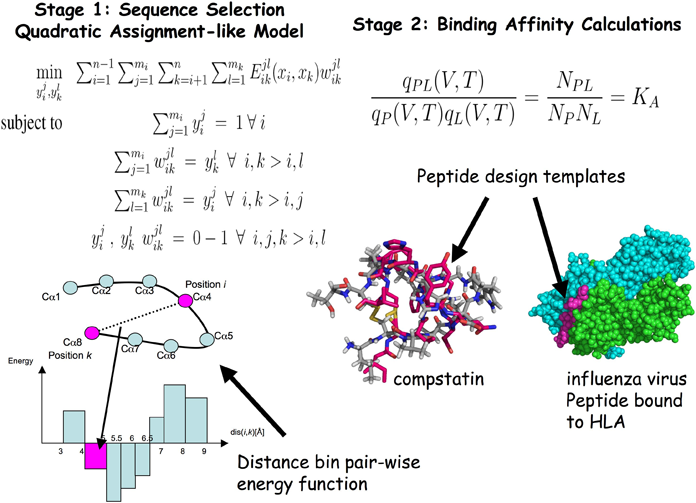
The figure illustrates the two aspects to the de novo protein design framework that has been developed in the Floudas research group. The left side of the figure illustrates stage one, the sequence selection stage. Stage one solves a quadratic assignment-like model to generate a rank-ordered list of amino acid sequences with the lowest energies that will fold into a given template structure. The energy of each sequence is calculated using a distance-dependent, pair-wise force field. The model incorporates the inherent flexibility of a protein in two ways: first by using a distance bin approach in the energy function, and second by utilizing multiple template structures as the design template. The right side of the figure illustrates stage two, the binding affinity calculation stage. For each amino acid sequence from stage one, the binding affinity of that peptide bound to a target protein is calculated using approximate partition functions (q) of the protein-peptide complex (PL), the peptide by itself (L), and the protein by itself (P). The ratio of these partition functions is related to the ratio of the concentrations of the respective proteins in solution, and therefore equal to its binding affinity, KA. The lower right portion of the figure shows illustrations of two peptide complexes: one for compstatin and another for an influenza virus peptide bound to HLA. The peptide templates are shown in pink. |