Research HighlightsStructure Prediction in Protein Folding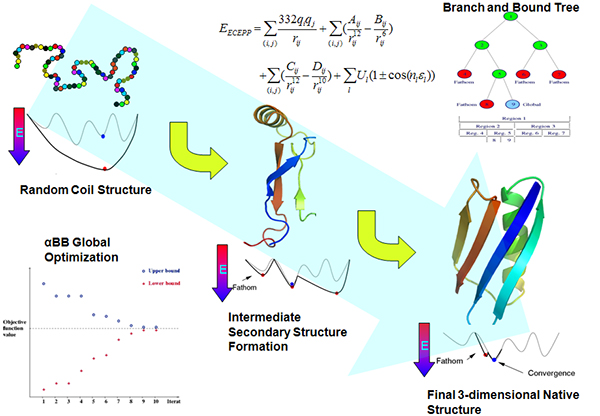
The protein folding problem is one of the most challenging and interesting problems in computational biology. The main aim is to predict the final three dimensional structure of the protein, given its amino acid sequence. The figure illustrates the hierarchical protein folding model, wherein the local structures (also known as protein secondary structure) are formed much faster compared to the final structure of the protein, which is formed by the final stabilizing interactions between the secondary structures. The final three-dimensional structure is believed to lie at its global free energy minimum. At the Floudas research group, we use αBB, a deterministic global free energy optimization framework to evaluate the final 3-d structure of the protein using ab initio techniques. The hierarchical protein folding process is carried out by initially predicting the secondary structure of a target protein. This is followed by predicting the topology, or the backbone fold, of the protein. This is used to finally predict the structure of the protein using deterministic global optimization and stochastic annealing. |